Creating places that enhance the human experience.

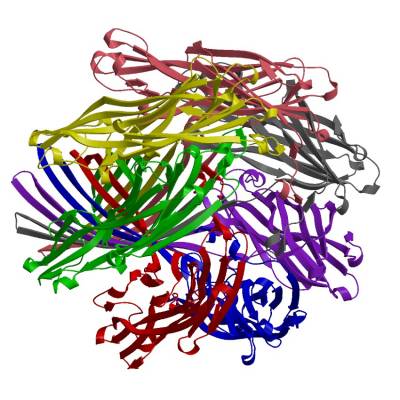
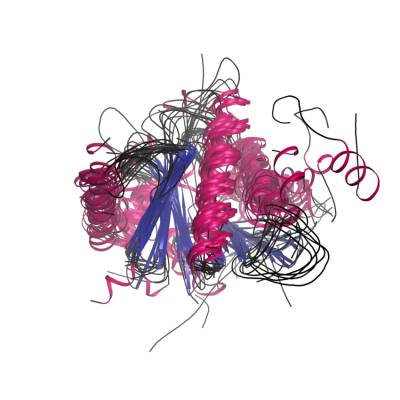
Evolution of protein families
We are researching the extent to which functions are conserved across families and in developing better methods for predicting when and how the functions of relatives change in different contexts. We have used our family classification to study the evolution of protein families and to perform comparative genomics. For example to determine which families are under or over represented in different organisms or different environments (e.g. in the metagenomics data).
Prediction of protein function
We have also developed methods for predicting protein associations and functional networks. Our methods for predicting functions and functional networks are being exploited by experimental groups in several EU funded collaborations in which we participate. These groups are researching proteins involved in cancer, angiogenesis, B-cell signalling and differentiation.
Collaborations
We also belong to the London Pain Consortium who are researching chronic pain. We participate in the NIH funded Protein Structure Initiative one of whose aims is to determine the structures of proteins of biological and medical interest.
Useful options
Beautiful snippets
Amazing pages
Building Molecular Tools
We are very pleased to announce that Prof Christine Orengo has been elected as a member of the European Molecular Biology Organisation (EMBO). EMBO is an organisation that promotes excellence across all aspects of the life sciences through courses, workshops, conferences and publications.






